A plagiarism story
The felony
On the 11/03/2014, I discovered that section 3.3.2 (pages 15 and 16) of a literature survey on Multi-Level Agent-Based Modeling that I update regularly on arXiv.org had been plagiarized by Ali Masoudi-Nejad and Alireza Meshkin from the University of Tehran in this paper, accepted for publication in Seminars in Cancer Biology.
Original
Cancer is a complex spatialized multi-scale process, starting from genetic mutations and potentially leading to metastasis. Moreover, it has multi-scale (from from genetic to environmental) causes. Therefore, it can be studied from various perspectives from the intracellular (molecular) to the population levels.ML-ABM is a promising paradigm to model cancer development and discover new therapies (Wang and Deisboeck, 2008, 2013). [...]
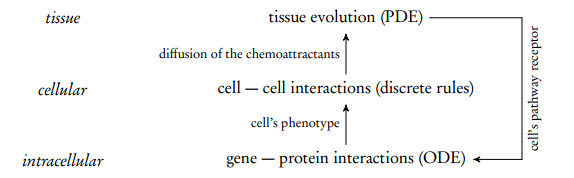
Zhang et al. developed an ML-ABM of a brain tumor named Glioblastoma Multiforme (GBM) (Zhang et al., 2007, 2009a, 2011, 2009b). This model explicitly defines the relations between scales and uses different modeling approaches: ordinary differential equations (ODE) at the intracellular level, discrete rules typically found in ABM at the cellular level and partial differential equations (PDE) at the tissue level (see fig. 6).
Moreover, this model also relies on a multi-resolution approach: heterogenous clusters, i.e., composed of migrating and proliferating cells are simulated at a high resolution while homogenous clusters of dead cells are simulated at a lower resolution. [...] This model has been implemented on graphics processing units (GPU), leading to an efficient parallel simulator (Zhang et al., 2011). Then, this model illustrates two interesting features of the ML-ABM approach: the coupling of heterogeneous models and the dynamic adaptation of the level of detail of simulations to save computational resources. [...]
Lepagnot and Hutzler (2009) model the growth of avascular tumors to study the impact of PAI-1 molecules on metastasis. To deal with the problem complexity (a tumor may be composed of millions of cells) two levels are introduced: the cell and the tumor’s core levels (fig. 6). Indeed, such cancers are generally structured as a kernel of necrosed or quiescent cells surrounded by living tumor cells. As necrosed and quiescent cells are mostly inactive, tumor’s core is reified as a single upper-level agent, interacting with cells and PAI-1 molecules at its boundary. A more comprehensive analysis of this model can be found in Gil-Quijano et al. (2012).
Figure 6:
Copy
Cancer is a complex specialized multi-scale process that can be studied from the intracellular, cellular or tissue perspectives. Therefore, agent-based modeling is a promising paradigm to model cancer development [21]. Zhang et al. developed an agent-based modeling of a brain tumor named Glioblastoma Multiforme (GBM) [2],[22], [23] and [24]. The different modeling approaches are used in this model: ordinary differential equations (ODE) at the intracellular level, discrete rules typically found in ABM at the cellular level and partial differential equations (PDE) at the tissue level (Fig. 2). Moreover, this model also relies on a multi-resolution approach: heterogeneous clusters, i.e. composed of migrating and proliferating cells are simulated at a high resolution while homogenous clusters of dead cells are simulated at a lower resolution. More computational resource is allocated to heterogenous regions of the cancer and less to homogenous regions [23]. This model incorporates a graphics processing unit (GPU)-based parallel computing algorithm [25], to speed up the previous agent-based models [22] and [24].Lepagnot and Hutzler, developed an agent-based system for modeling the growth of avascular tumors to study the impact of PAI-1 molecules on metastasis [26]. To deal with the problem of complexity (a tumor may be composed of millions of cells) two levels are introduced: the cell and the tumor's core levels. Indeed, such cancers are generally structured as a kernel of necrosed or quiescent cells surrounded by living tumor cells. As necrosed and quiescent cells are mostly inactive, tumor's core is reified as a single upper-level agent, interacting with cells and PAI-1 molecules at its boundary. A more comprehensive analysis of this model can be found in [27].
Figure 2:
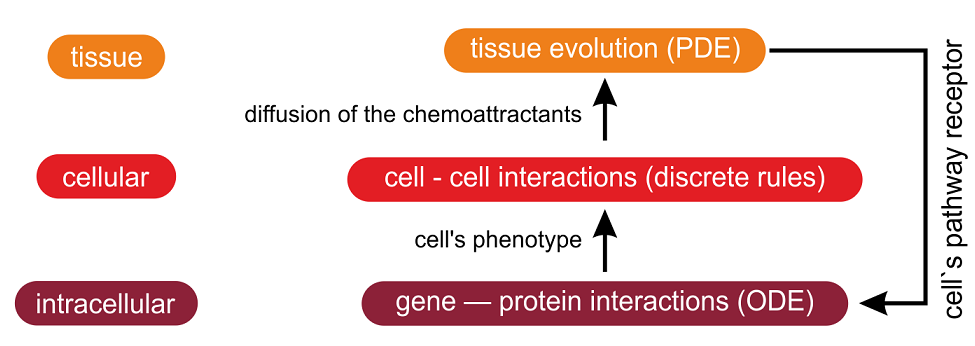
The references (except [25] which is another paper by Zhang et al. on the same subject) and the figures (numbered 6 in mine and 2 in the Seminars in Cancer Biology article) are the same. I also note that my own review is not cited.
I did an analysis with a plagiarism detection software. Results show significant text overlaps with other papers.
Check this annotated copy of the article to see the extent of the plagiarism.
This article has not been written but assembled from other texts: it is solely based on the copy/paste technique (with a little rewriting I must admit). Some plagiarized texts are cited but not all.
A onetime mistake or a bad habit?
But, is this a onetime mistake or a bad habit? A silimar analysis, contucted on a recent article of Ali Masoudi-Nejad and Alireza Meshkin shows that, unfortunaltely the correct answer is the latter. Indeed, it includes content from this paper, without citing it.
Update 14/09/2015: This paper has been finally retracted by the Editor-in-Chief of Mol. Genet. Genomics However, it is still possible to buy the paper for 43.69 €...
Original
There exists a wide range of disease gene prioritization methods that are based on the analysis of the topological properties of PPI networks. These methods commonly rely on the observation that the products of genes that are associated with similar diseases have a higher likelihood of physically interacting [11]. It is important to note here the distinction between genes and their products. Genome-wide association studies focus on identifying genes that are associated with a disease of interest. Network-based prioritization aims to aid this effort by inferring functional associations between genes based on the interactions among their products, i.e., proteins. For this reason, any reference to interactions between genes in this paper refers to the interactions between their products.Existing methods for network-based disease gene prioritization can be classified into two main categories; (i) localized methods, i.e., methods based on direct interactions and shortest paths between known disease genes and candidate genes [3,9,16], (ii) global methods, i.e., methods that model the information flow in the cell to assess the proximity and connectivity between known disease genes and candidate genes. Several studies show that global approaches, such as random walk and network propagation, clearly outperform local approaches [13,14,17]. For this reason, we focus on global methods in this paper.
For a given disease of interest D, the input to the disease gene prioritization problem consists of two sets of genes, the seed set S and the candidate set C. The seed set S specifies prior knowledge on the disease, i.e., it is the set of genes known to be asso- ciated with D and diseases similar to D. Each gene v ∈ S is also associated with a simi- larity score s(v, D), indicating the known degree of association between v and D. The similarity score for gene v is computed as the maximum phenotypic similarity between D and any other disease associated with v, based on clinical description of diseases (a detailed discussion on computation of phenotypic similarity scores can be found in the Methods section below). The candidate set C specifies the genes, one or more of which are potentially associated with disease D (e.g., these genes might lie within a linkage interval that is identified by association studies). The overall objective of network based disease prioritization is to use a human PPI network G = (V , ε, w), to compute a score a(v, D) for each gene v ∈ C, such that a(v, D) represents the likelihood of v to be associated with D.
The PPI network G = (V,ε,w) consists of a set of gene products V and a set of undirected interactions E between these gene products, where uv ∈ E represents an interaction between u ∈ V and v ∈ V. Since PPI data might be obtained from various resources, interactions are often assigned confidence scores indicating their reliability. In other words, the network is also associated with a function w : E → (0, 1], such that w(uv) indicates the reliability of interaction uv ∈ E. Finally, the set of interacting part- ners of a gene product v∈V is defined as N(v) ={u∈V:uv∈ε }and the total reliability of known interactions of v is defined as W(v) = u∈N(v) w(uv) which we refer as weighted degree throughout this paper. Global prioritization methods use this network information to compute a by propagating s over G. Candidate proteins are then ranked according to a and novel genes that are potentially associated with the disease of interest are identified based on this ranking.
etc.
Copy
A wide range of methods based on the analysis of the topological properties of PPI networks exist. These methods commonly rely on the expectation that the products of the genes that are associated with similar diseases interact heavily with each other. It is important to note that the purpose here is to infer associations between genes from functional and physical interactions between their products. For this reason, any reference to the interactions between genes in this paper necessarily refers to their protein or RNA products. Existing methods can be classified into two main categories; (1) localized methods, i.e., methods based on direct interactions and shortest paths between known disease genes and candidate genes (Lage 2007; Oti et al. 2006; George et al. 2006) (2) global methods, i.e., methods that model the information flow in the cell to assess the proximity and connectivity between known disease genes and candidate genes. Several studies show that global approaches, such as random walk and network propagation, clearly outperform local approaches (Kohler et al. 2008; Vanunu and Sharan 2008).For a given disease of interest D, the input to the can- didate disease gene prioritization problem consists of two sets of genes, seed set S and candidate set C. The seed set S specifies prior knowledge on the disease, i.e., it is the set of genes known to be associated with D and diseases similar to D. Each gene v ∈ S is also associated with a similarity score r(v, D), indicating the known degree of association between v and D. The similarity score for gene v is com- puted as the maximum similarity between D and any other disease associated with v. The candidate set C specifies the genes, one or more of which are potentially associated with disease D (e.g., these genes might lie within a linkage interval that is identified by association studies). The overall objective of this network-based disease prioritization is to use a human PPI network G = (V, E), to compute a score a(v, D) for each gene v ∈ C that represents the likelihood of v to be associated with D.
The PPI network G = (V, E) consists of a set of gene products V and a set of undirected interactions E between these gene products where uv ∈ E represents an interaction between u ∈ V and v ∈ V. In this network, these tof interacting partners of a gene product v ∈ V is defined as N(v) = {u ∈ V : uv ∈ E}. Global prioritization schemes use this network information to compute a by propagating r over G. Candidate proteins are then ranked according to a and novel genes that are potentially associated with the disease of interest are identified based on this ranking.
etc.
And I did not reproducted the whole plagiarized text!
You want more? Compare this paper and this one.
Conclusion
Then, while Ali Masoudi-Nejad denies it, I think that this article constitutes an obvious, deliberate and methodical case of plagiarism.
Following my complaint, the editor of Seminars in Cancer Biology informed the corresponding author and I on the 18/03/2014 that the paper would be withdrawn. On the 04/04/2014, it was not possible to buy or access the article on sciencedirect anymore. Unfortunately, the article page does not say why it has been withdrawn...
Last edited: 08/02/2016